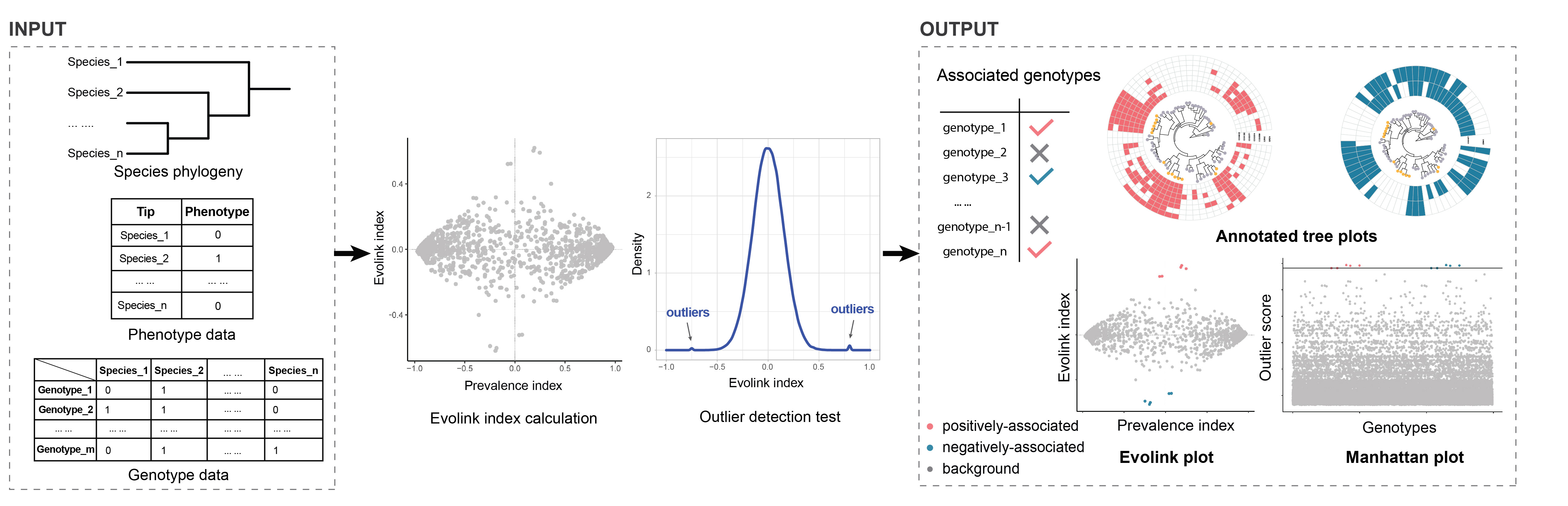

Prepare your input files and name them 'tree.nwk', 'gene.tsv' and 'trait.tsv' for your species tree, genotype matrix and phenotype list, respectively.
Details about the format of these files can be found at About Page and the rest of this page.
The names should be in accordance with what has been shown here, otherwise, the tool will not recognize the inputs.
Since files can be very large, we highly recommend users to compress them into a zip file.
Note that before compression, don't zip any folder. Please select these input files and directly zip them.
A zipped sample input is also provided. Feel free to download it and make a test.
Newick format tree file. It is recommended that the tree is rooted. Internal node names are not necessary. For example:
(species_1:1,(species_2:1,(species_3:1,species_4:1)Internal_1:0.5)Internal_2:0.5)Root:0.1;
Trait/Phenotype binary file (tab separated file). The header is a must and should be "Tip" and "Status". Tip column contains the tip names the same as the tree, while Status column contains the presence (1) and absence (0) of the phenotype for each leaf. So far only 1 or 0 is accepted and all leaves should be labeled with a 0/1 status. For example:
| Tip | Status |
|---|---|
| species_1 | 0 |
| species_2 | 1 |
| species_3 | 1 |
| species_4 | 0 |
Gene presence/absence matrix file (tab separated file). Each row is the binary (0/1) status of each gene across all species. Each gene should appear in a species for at least one time. The first colname could be any word, but "orthoID" (orthogroup ID) is a nice choice to be shown here. For example:
| OrthoID | species_1 | species_2 | species_3 | species_4 |
|---|---|---|---|---|
| gene_1 | 0 | 1 | 1 | 0 |
| gene_2 | 1 | 1 | 0 | 0 |
| gene_3 | 1 | 0 | 1 | 0 |
| gene_4 | 0 | 1 | 0 | 1 |

Alternatively, you can upload the input files separately. In this case, you can name the files whatever you like. But pay attention to the file extension since only files ending with ".tsv" or ".nwk" are allowed.
A zipped sample input is provided. Uncompress it to get three input files before test.
This information is optional. If a job name is provided, you could see it on the output page. Otherwise, "NoJobName" will be automatically assigned to your job.
If the input is big, we recommend the user to provide their emails, so that they don't need to keep the running window open and the result will be sent to the user's email address once the job is done.
Better to double-check and make sure you provide a correct email address.
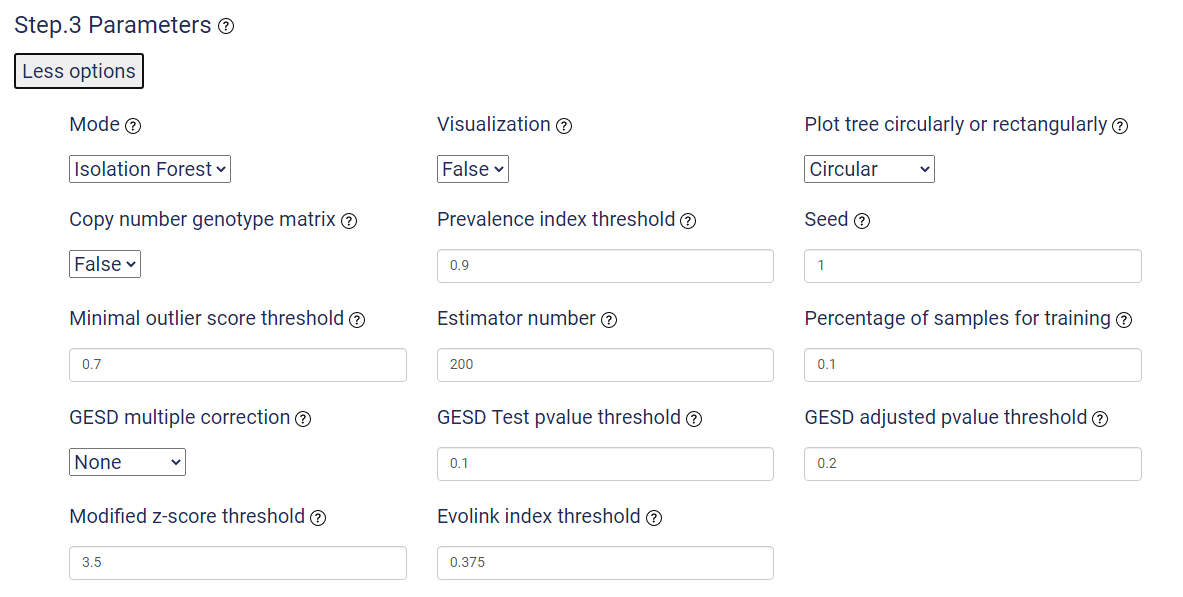
By clicking "More options", more parameters will show. By default, the "Visualization" option is "False" to save running time and switch it on if you need to visualize your results.
| Parameter | Description |
|---|---|
| Mode | Evolink has four modes insofar to detect phenotype-assoicated genotypes: isolation_forest, gesd_test,(modified) z_score, and cutoff. Choices: isolation_forest, gesd_test, z_score, cutoff. Default: isolation_forest |
| Visualization | Whether to generate plots for visualization. Three types of plots will be generated: Evolink plot, Manhattan plot and tree plot annotated with the prsence/absence of the top 5 positively and negatively assoicated genotypes. |
| Plot tree circularly or rectangularly | If this option is True, the tree plot can be displayed either circularly or rectangularly. |
| Copy number genotype matrix | If this option is True, the given genotype matrix includes continuous genotypes (e.g. gene copy numbers) instead of presence/absence binary values. And Evolink will internally convert the continuous values into binary values. |
| Prevalence index threshold | Absolute threshold used to filter prevalent and rare genotypes. Only works for gesd_test and z_score modes. |
| Seed | Set seed for reproducibility of the results. |
| Minimal Outlier score threshold | A minimal threshold to determine outliers in isolation_forest mode. Isolation forest mode detects the ourlier score threshold by finding the maximal difference between outlier scores. But if the threshold is lower than a minimal value, this parameter will be used as the threshold instead. The aim of this parameter is to set a lower boundary and avoid finding too many outliers. Default=0.7. Range=0.5-1. |
| Estimator number | Number of tree estimators used in isolation_forest mode. |
| Percentage of samples for training | Maximal percentage of training samples for each tree used in isolation_forest mode. Default=0.1. Range=0-1. |
| GESD multiple correction | Multitest correction method for p-values in gesd_test mode. Choices: none, bonferroni, fdr, holm, hommel. Default=none. |
| GESD Test pvalue threshold | Threshold for original p-values in gesd_test mode. Default=0.1. |
| GESD adjusted pvalue threshold | Threshold for adjusted p-values in gesd_test mode if GESD multiple correction option is not "none". Default=0.2. |
| Modified z-score threshold | Absolute modified z-score threshold used in z_score mode. Default=3.5. Usually, 2 corresponds to top 97.73% of the distribution, 2.5 to 99.38%, 3 to 99.87% and 3.5 to 99.98%. |
| Evolink index threshold | Absolute Evolink index threshold used in cutoff mode to select significant genotypes. Default=0.375. |
You can also directly click "run the example data" to simply test the above sample input and quickly check how Evolink webserver works.
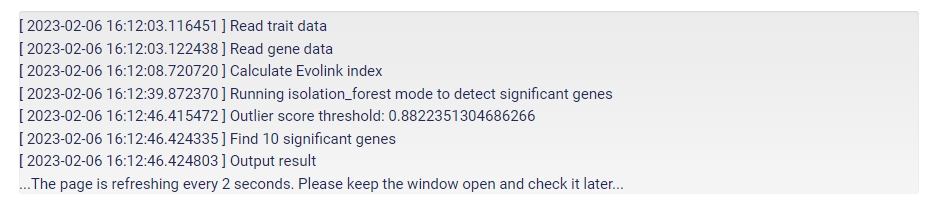
After clicking "submit" and uploading the input files, your job status will be updated every 2 seconds. To get a result, please don't close the window.
If you dislike keeping the window open, remember to provide your email address before submission so that you could still get the results from email.
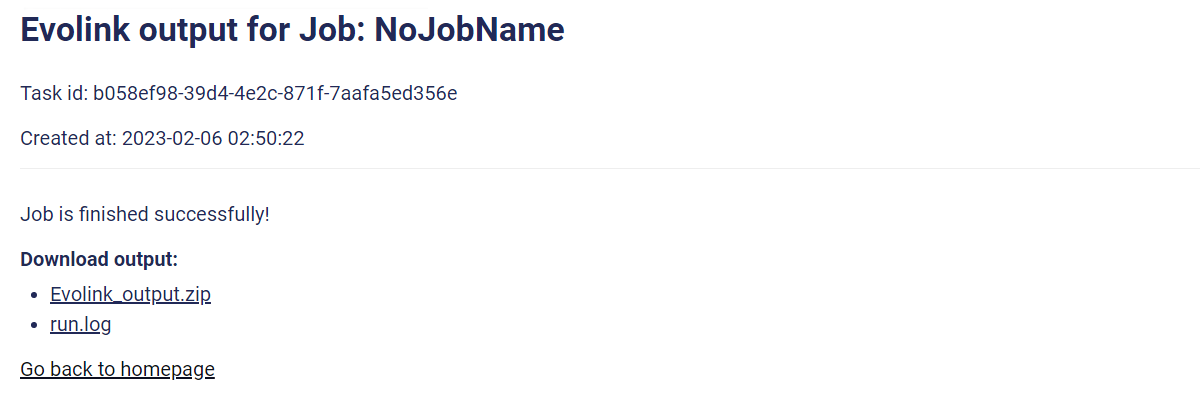
Thanks for your patience. If your job is done successfully, you can download the result and the running log file as shown above.
If job fails, please download the log file and check what goes wrong.